










FDA批准艾伯维依鲁替尼联合利妥昔单抗治疗罕见淋巴瘤
时间:2018-08-29 作者:联创生物医药信息部
新浪医药
艾伯维(AbbVie)8月27日宣布,美国FDA批准了IMBRUVICA®(依鲁替尼,Ibrutinib)联合利妥昔单抗(RITUXAN®)治疗成人特发性巨球蛋白血症(WM),这是一种罕见且无法治愈的非霍奇金淋巴瘤(NHL)。 此次批准,意味着第一种也是唯一一种特别针对该疾病的无化疗联合疗法上市。
依鲁替尼于2015年1月作为单药治疗WM首次被批准,目前共获得九项FDA批准,涵盖六种不同的疾病。该药物是由艾伯维旗下的Pharmacyclics公司和杨森生物公司共同开发和商业化的一种首创布鲁顿氏酪氨酸激酶(BTK)抑制剂。无论是联合利妥昔单抗或是作为单药使用,成人WM患者IMBRUVICA推荐剂量为每日口服420mg一次直至出现疾病进展或不可接受的毒性。当IMBRUVICA联合利妥昔单抗给药时,需考虑同一天在利妥昔单抗给药之前给药IMBRUVICA。
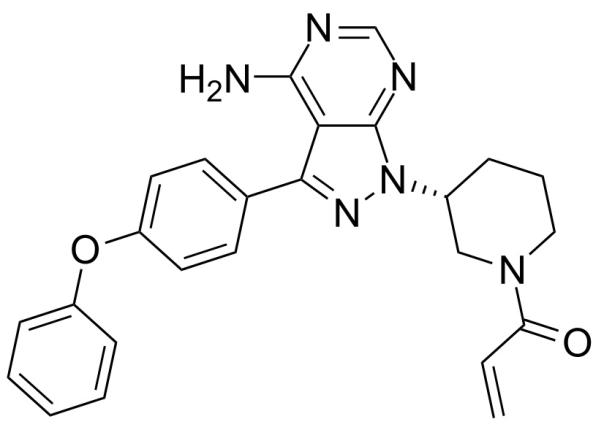
Ibrutinib分子结构(来自维基百科)
艾伯维旗下Pharmacyclics公司的临床开发主管Thorsten Graef医学博士说,“我们很高兴不管是作为单药还是联合利妥昔单抗,依鲁替尼都获得了批准,为患有特发性巨球蛋白血症的患者提供了另外的有效治疗选择。我们为公司强劲的临床开发项目而感到自豪,这一新的批准也反映了我们不断致力于探索依鲁替尼作用机理的全部潜能,来治疗那些未满足医疗需要的疾病患者。”
此项新的FDA批准是基于临床3期iNNOVATE(PCYC-1127)试验的数据。该试验通过与单独使用利妥昔单抗对比,在150例之前未治疗以及复发/难治性WM患者中评估了依鲁替尼联合利妥昔单抗的药效。在为期26.5个月的中位随访中,该研究显示,与单独使用利妥昔单抗相比,依鲁替尼联合利妥昔单抗的患者无进展生存期(PFS)显著改善(30个月PFS率分别为82%和28%);且其疾病进展或死亡的相对危险度也降低了80%(风险比0.20;置信区间:0.11-0.38,P 》上。
iNNOVATE是一项由Pharmacyclics公司赞助的随机、安慰剂对照、双盲临床3期研究,患者随机接受静脉滴注利妥昔单抗375 mg/m2,每周一次,连续4周,紧接着是第二次为期3个月,每周四次的利妥昔单抗注射疗程。所有患者每天接受一次420mg依鲁替尼或安慰剂,直至达到永久停药的标准。主要终点是无进展生存期,而作为每个治疗组中衡量安全性和耐受性标准的次要终点包括总体反应率、通过血红蛋白测量的血液改善、到下次治疗时间、总生存期以及出现不良事件的受试人数。该研究中,所有不同形式的最常见不良反应(发生在20%或更多患者中)有淤伤(37%)、肌肉骨骼疼痛(35%)、出血(32%)、腹泻 (28%)、皮疹(24%)、关节痛(24%)、恶心(21%)和高血压(20%)。
雅典大学医学院临床治疗学系教授和*、带领iNNOVATE研究的Meletios A. Dimopoulos博士说,“该研究中令人信服的临床证据支持了IMBRUVICA联合利妥昔单抗在治疗特发性巨球蛋白血症中的疗效。此项批准对于没有其他治疗选择的WM患者群体来说是一个重要的里程碑。”
Dana-Farber癌症研究所特发性巨球蛋白血症研究中心主任、哈佛医学院副教授、IMBRUVICA临床2期试验领头研究者Steven P. Treon医学博士说,“依鲁替尼已显著推进了特发性巨球蛋白血症的治疗,该药联合利妥昔单抗的批准,无疑为许多WM患者增加了一个新的治疗选择。”
特发性巨球蛋白血症是一种罕见的、缓慢生长且无法治愈的非霍奇金淋巴瘤,治疗方案有限,该病通常影响中老年人,尽管淋巴结和脾也可能受到影响,但主要发现于骨髓中。在美国,每年约有2,800例新病例。
【版权提示】 联创生物医药发布原文章与转载文章。转载文章的目的是推荐有价值的信息,与更多的朋友分享,我们尊重知识产权,致力于保护知识产权,尽力注明作者和出处。如果我们转载的文章、图片等内容存在版权等问题,敬请联系我们0551-68596228,我们将尽全力尽快与您联系,及时处理。衷心感谢您的理解与惠助!

